王立堃研究组发现未折叠蛋白响应调控外泌体分泌的新机制
发布时间:2025-07-28
细胞的囊泡转运在蛋白质、脂质等物质的转运和分泌中起到重要作用。经典的蛋白分泌途径由内质网-高尔基体介导,但近年来不断发现新的非经典分泌途径,外泌体即为其中之一,这是由细胞内一种特殊的囊泡-多囊泡体(multivesicular body, MVB)与质膜融合后向胞外释放的产物,通过物质的跨细胞转运对生命活动产生广泛影响。外泌体的产生和分泌与细胞应激状态密切相关,比如在经典分泌途径受影响的内质网应激条件下,外泌体的释放会增加。细胞在感受到内质网应激时,是如何启用外泌体这一非经典分泌途径的呢?
2025年7月21日,中国科学院生物物理研究所王立堃团队在《Journal of Biological Chemistry》在线发表了题为"PERK and IRE1α promote exosome secretion via blocking lysosomal degradation of multiple vesicular body"的研究论文。该研究发现,内质网应激诱发的细胞未折叠蛋白响应(unfolded protein response, UPR)信号通路PERK和IRE1α能够降低溶酶体酸度,抑制MVB和溶酶体的融合,从而减少溶酶体对MVB的降解,促进小鼠黑色素瘤细胞的外泌体分泌。
首先,研究团队利用内质网应激的诱导剂毒胡萝卜素(Thapsigargin, Tg)激活UPR,发现敲除IRE1α显著降低细胞外囊泡(Extracellular Vesicles, EVs)分泌,而敲除PERK对EVs分泌没有明显影响,蛋白质组分析表明分离到的EVs主要来源于内吞途径,因此可被认为是外泌体。此外,敲除PERK会导致IRE1α下游的XBP1s蛋白显著上调,这表明在内质网应激条件下,IRE1α可能补偿PERK对外泌体分泌的影响。因此,研究团队利用IRE1α抑制剂KIRA8预处理细胞,发现当IRE1α被抑制时,PERK缺失减少了外泌体分泌。以上结果表明,在内质网应激条件下,PERK和IRE1α通路都有助于外泌体分泌。
接下来,研究团队发现在内质网应激和IRE1α缺陷细胞中,PERK缺失增加溶酶体酸度,增强溶酶体活性,促进MVB和溶酶体融合,导致MVB降解,从而抑制外泌体分泌。进一步地,研究团队发现PERK通路下游的转录因子ATF4是参与调节溶酶体活性和外泌体分泌的关键因子。ATF4通过抑制溶酶体膜上质子泵V-ATPase的组装下调溶酶体酸化。研究团队前期发现胞浆Ca2+能特异结合并上调PERK活性(原文链接:https://www.ncbi.nlm.nih.gov/pubmed/36870060)。受此启发,他们使用离子霉素(ionomycin)处理细胞以提高胞质Ca2+浓度来特异性激活PERK,发现该处理在不引起内质网应激的条件下即可诱导外泌体分泌,这表明PERK激活可以独立于内质网应激促进外泌体产生。
最后,在IRE1α调控外泌体分泌方面,研究团队发现敲除IRE1α同样通过增加溶酶体酸度,增强溶酶体活性从而抑制外泌体分泌。具体的分子机制还有待进一步阐明。
综上所述,这项研究揭示了UPR信号通路PERK和IRE1α调控外泌体分泌的机制,表明源自内质网的UPR信号可以调节囊泡转运过程中的降解-分泌的平衡。
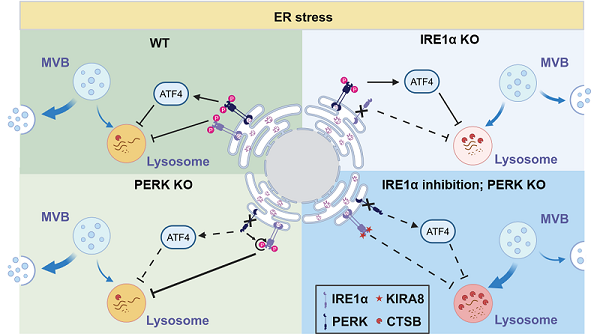
图:左上,在内质网应激条件下,UPR下游的IRE1α和PERK激活,减弱溶酶体酸度,降低溶酶体活性,抑制MVBs和溶酶体融合,促进外泌体分泌。右上,在内质网应激条件下,IRE1α缺失提高溶酶体酸度,增强溶酶体活性,从而抑制外泌体分泌。左下,在内质网应激条件下,PERK缺失导致IRE1α补偿性激活,补偿了PERK缺失对外泌体分泌的影响。右下,在内质网应激和IRE1α缺陷细胞中,PERK缺失进一步提高溶酶体酸度,增强溶酶体活性,促进MVBs和溶酶体融合,导致MVBs降解,从而抑制外泌体分泌。
中国科学院生物物理研究所王立堃研究员为该论文的通讯作者,已毕业博士周仕新为论文的第一作者。该研究 受到国家自然科学基金、国家重点研发计划、中国科学院战略性先导科技专项以及中国科学院青年交叉团队项目的资助。
文章链接:
https://www.jbc.org/article/S0021-9258(25)02354-3/fulltext
院英文网报道链接:
https://english.cas.cn/newsroom/research_news/life/202508/t20250804_1048979.shtml
(供稿:王立堃研究组)
附件下载: